Seqenv 1.3.0
seqenv
- Assign environment ontology (EnvO) terms to short DNA sequences.
- All code written by Lucas Sinclair.
- Publication at: https://peerj.com/articles/2690/
Usage
Once that is done, you can start processing FASTA files from the command line. For using the default parameters you can just type:
$ seqenv sequences.fasta
We will then assume that you have inputed 16S sequences. To modify the database or input a different type of sequences:
$ seqenv sequences.fasta --seqtype prot --search_db nr
To modify the minimum identity in the similarity search, use the following:
$ seqenv sequences.fasta --min_identity 0.97
If you have abundance data you would like to add to your analysis you can specify it like this in a TSV file:
$ seqenv sequences.fasta --abundances counts.tsv
All parameters
Several other options are possible. Here is a list describing them all:
--seq_type: Sequence typenuclorprot, for nucleotides or amino acids respectively (Default:nucl).--search_algo: Search algorithm. Eitherblastorvsearch(Default:blast).--search_db: The database to search against (Default:nt). You can specify the full path or make a~/.ncbircfile.--normalization: Can be either offlat,uiorupui. This option defaults toflat.- If you choose
flat, we will count every isolation source independently, even if the same text appears several times for the same input sequence. - If you choose
ui, standing for unique isolation, we will count every identical isolation source only once within the same input sequence. - If you choose
upui, standing for unique isolation and unique pubmed-ID, we will uniquify the counts based on the text entry of the isolation sources as well as on the pubmed identifiers from which the GI obtained.
- If you choose
--proportional: Should we divide the counts of every input sequence by the number of envo terms that were associated to it. Defaults toTrue.--backtracking: For every term identified by the tagger, we will propagate frequency counts up the acyclic directed graph described by the ontology. Defaults toFalse.--restrict: Restrict the output to the descendants of just one ENVO term. This removes all other terms that are not reachable through the given node. For instance you could specify:ENVO:00010483(Disabled by default)--num_threads: Number of cores to use (Defaults to the total number of cores). Use1for non-parallel processing.--out_dir: The output directory in which to store the result and intermediary files. Defaults to the same directory as the input file.--min_identity: Minimum identity in similarity search (Default:0.97). Note: not available when usingblastp.--e_value: Minimum e-value in similarity search (Default:0.0001).--max_targets: Maximum number of reference matches in the similarity search (Default:10).--min_coverage: Minimum query coverage in similarity search (Default:0.97).--abundances: Abundances file as TSV with OTUs as rows and sample names as columns (Default: None).--N: If abundances are given, pick only the top N sequences (Disabled by default).
Why make this ?
The continuous drop in the associated costs combined with the increased efficiency of the latest high-throughput sequencing technologies has resulted in an unprecedented growth in sequencing projects. Ongoing endeavors such as the Earth Microbiome Project and the Ocean Sampling Day are transcending national boundaries and are attempting to characterize the global microbial taxonomic and functional diversity for the benefit of mankind. The collection of sequencing information generated by such efforts is vital to shed light on the ecological features and the processes characterizing different ecosystems, yet, the full knowledge discovery potential can only be unleashed if the associated meta data is also exploited to extract hidden patterns. For example, with the majority of genomes submitted to NCBI, there is an associated PubMed publication and in some cases there is a GenBank field called “isolation sources” that contains rich environmental information.
With the advances in community-generated standards and the adherence to recommended annotation guidelines such as those of MIxS of the Genomics Standards Consortium, it is now feasible to support intelligent queries and automated inference on such text resources.
The Environmental Ontology (or EnvO) will
be a critical part of this approach as it gives the ontology for the concise,
controlled description of environments. It thus provides structured and
controlled vocabulary for the unified meta data annotation, and also serves as
a source for naming environmental information. Thus, we have developed the
seqenv pipeline capable of annotating sequences with environment descriptive
terms occurring within their records and/or in relevant literature.
The seqenv pipeline can be applied to any set of nucleotide or protein
sequences. Annotation of metagenomic samples, in particular 16S rRNA sequences
is also supported.
The pipeline has already been applied to a range of datasets (e.g Greek lagoon, Swedish lake/river, African and Asian pitlatrine datasets, Black Sea sediment sample datasets have been processed).
What does it do exactly ?
Given a set of DNA sequences, seqenv first retrieves highly similar
sequences from public repositories (e.g. NCBI GenBank) using BLAST or similar
algorithm. Subsequently, from each of these homologous records, text fields
carrying environmental context information such as the reference title and the
isolation source field found in the metadata are extracted. Once the
relevant pieces of text from each matching sequence have been gathered, they
are processed by a text mining module capable of identifying any EnvO terms
they contain (e.g. the word “glacier”, or “pelagic”, “forest”, etc.). The
identified EnvO terms along with their frequencies of occurrence are then
subjected to multivariate statistics, producing matrices relating your samples
to their putative sources as well as other useful outputs.
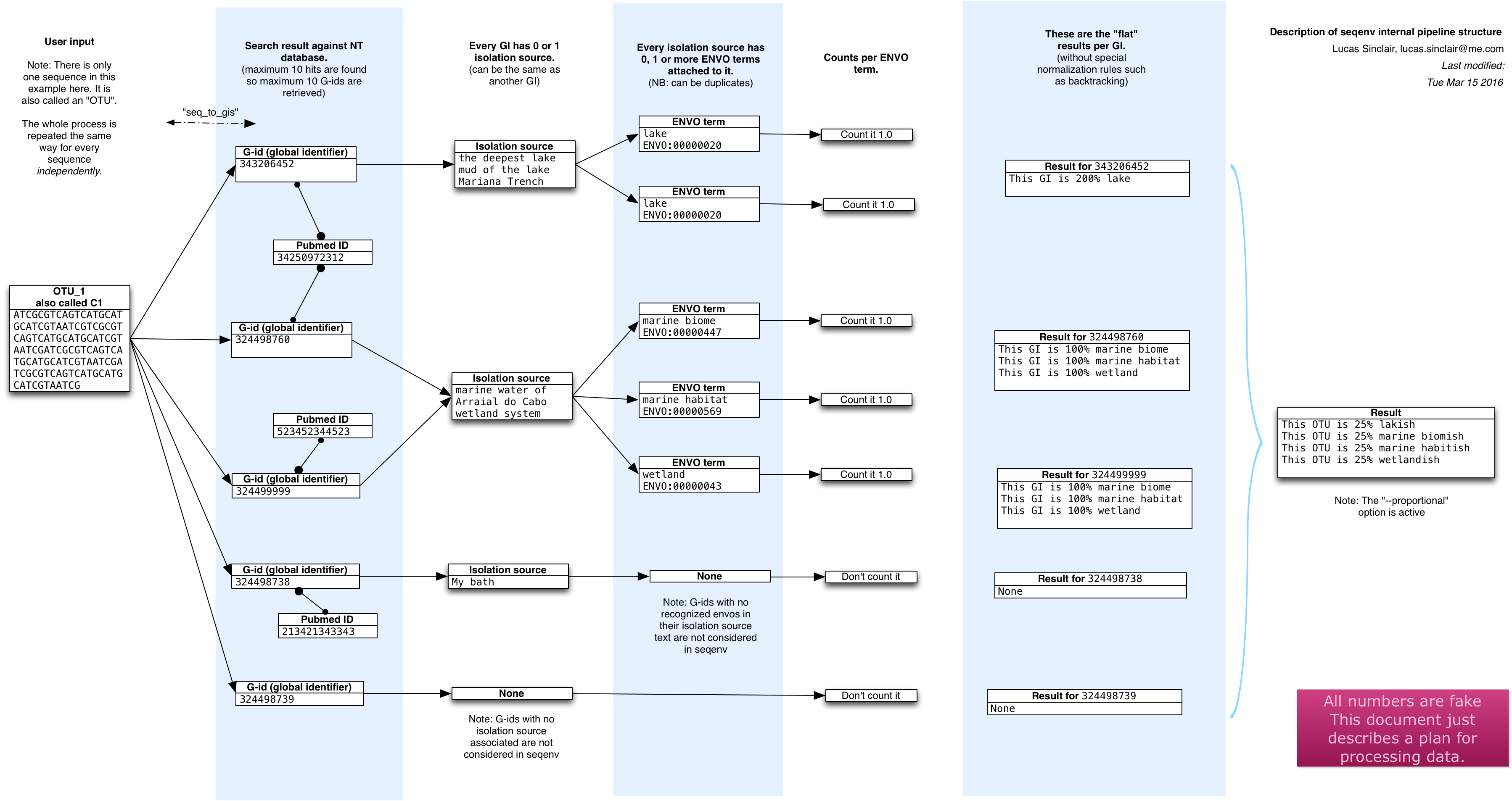
Pipeline overview
The publication contains more information of course, but here is a schematic
overview of what happens inside seqenv:
Tutorial
We will first run seqenv on a 16S rRNA dataset using isolation sources
as a text source. Here, abundance.tsv is a species abundance file (97% OTUs)
processed through illumitag software
and centers.fasta contains the corresponding sequences for the OTUs.
|
|
The output you will receive should look something like this:
|
|
Once the pipeline has finished processing, you will have the following contents in the output folder:
|
|
The most interesting files are probably:
list_concepts_found.tsvlinks every OTU to all its relevant BLAST hits and linked ENVO terms.seq_to_names.tsva matrix linking every OTU to its “composition” in terms of ENVO identifiers translated to readable names.samples_to_names.tsvif an abundance file was provided, this is a a matrix linking every one of your samples to its “composition” in terms of ENVO identifiers translated to readable names.graphviz/directory containing ontology graphs for everyone of the inputed sequences, such as in the following example:

Activating the conda environment
Check out a node with qrsh and run:
|
|
To use over SGE, add the source line above to your shell scripts prior to your seqenv commands.
Usage notes
Please always set --num_threads to the desired number of threads, otherwise
all available threads will be used.
The seqenv command is looking for a specific version of nt. This version
is already downloaded in /nfs1/CGRB/databases/seqenv so you do not need to
download it. The $BLASTDB variable will update upon conda env activation, so
you can reference it using nt (or just leave the db as default).
Location and version
|
|
help message
|
|
software ref: https://github.com/xapple/seqenv
research ref: https://doi.org/10.7717/peerj.2690